BIO520 Final Exam Spring 2010
Please email this lab to Jim Lund (jiml@uky.edu) with a subject line "BIO520 Final Exam" and name the document like so: "LundJ_exam2" or hand in written answers. Fill in your name on the exam!
You may use any books, notes, web pages, software programs, or related materials to complete this exam. You MAY NOT consult with any person regarding the exams intellectual content.
1. Genbank
- a (1 pt). Find the RefSeq protein entry for human CYP3A4, a cytochrome P450. Give the RefSeq accession number.
- b (1 pt). Where is this gene located in the human genome? Give the chromosome and approximate location in Mb. Example: Ch2, 13.4 Mb.
- c (2 pts). What genes are adjacent to CYP3A4 on the chromosome?
2. Examine the PDB entry 1W0E for CYP3A4.
- a (1 pt). What method was used to determine this structure, and what is its resolution?
- b (1 pt). What is the large molecule in the center of the protein?
- c (2 pt). Examine the Ramachandran plot for this structure: 1W0E_Rama.pdf. What does the plot tell you about the quality of the structure? Explain your answer.
3. Human sirtuin 1 protein was BLASTed against the nr database. A one BLAST match is shown below.
- a (1 pt). Examine the HSP shown below. What do the ‘---‘ characters in the query sequence at aa 220-237 indicate?
- b (1 pt). Examine the HSP shown below. What is the difference between ‘Identities’ and ‘Positives’?
- c (1 pt). Examine the HSP shown below. Does the E-value for this HSP indicate this is a excellent, borderline, or insignificant match?
- d (1 pt). Examine the HSP shown below. Would you expect a BLASTN comparison of the DNA sequences for these two proteins to show higher or lower percent identical bps?
>ref|XP_635962.1| Gene info NAD(+)-dependent deacetylase, silent information regulator protein (Sir2) family protein [Dictyostelium discoideum AX4]
Length=542
Score = 277 bits (708), Expect = 2e-72, Method: Composition-based stats.
Identities = 149/333 (44%), Positives = 211/333 (63%), Gaps = 31/333 (9%)
Query 185 YTFVQQHLMIGTDPRTILKDLLPETIPPPELD-DMTLWQIVINILSEP------------ 231
Y +Q+ +G DP KD+ + EL+ D W+I+ L+
Sbjct 215 YKHIQEKKSLGIDPIEFTKDIGFKL----ELEKDDDAWEIITAFLTRKKVAVNLFLNYLK 270
Query 232 ------PKRKK--RKDINTIEDAVKLLQECKKIIVLTGAGVSVSCGIPDFRSRDGIYARL 283
P RKK D++T E +L + K I+++TGAGVSVSCGIPDFRS+ G+Y +
Sbjct 271 YNTLARPYRKKIATLDLSTFEKVCQLFESSKNIVIITGAGVSVSCGIPDFRSKGGVYETI 330
Query 284 AVDFPDLPDPQAMFDIEYFRKDPRPFFKFAKEIYPGQFQPSLCHKFIALSDKEGKLLRNY 343
+ +LP P+++FDI Y R +P PFF+FAKEI+PG +PS H FI L D++GKLLRNY
Sbjct 331 EKKY-NLPRPESLFDIHYLRANPLPFFEFAKEIFPGNHKPSPTHSFIKLLDEKGKLLRNY 389
Query 344 TQNIDTLEQVAGIQR--IIQCHGSFATASCLICKYKVDCEAVRGDIFNQVVPRCPRCPAD 401
TQNIDTLE VAGI R ++ CHGSF+TA+C+ CK VD +R I +P C +C +
Sbjct 390 TQNIDTLEHVAGIDREKLVNCHGSFSTATCITCKLTVDGTTIRDTIMKMEIPLCQQC--N 447
Query 402 EPLAIMKPEIVFFGENLPEQFHRAMKYDKDEVDLLIVIGSSLKVRPVALIPSSIPHEVPQ 461
+ + MKP+IVFFGENLP++F + + D ++DLLIV+GSSL+V+PV+L+P + ++PQ
Sbjct 448 DGQSFMKPDIVFFGENLPDRFDQCVLKDVKDIDLLIVMGSSLQVQPVSLLPDIVDKQIPQ 507
Query 462 ILINREPLPHLH-FDVELLGDCDVIINELCHRL 493
ILINRE + H FD LGDCD + +L +++
Sbjct 508 ILINRELVAQPHEFDYVYLGDCDQFVQDLLNKV 540
4. BLAST
- a. (2 pts) The BLAST algorithm uses word matches between sequences to seed alignments. Megablast uses a longer word than blastn. How does this difference affect 1) the BLAST search and 2) the matches found by BLAST?
- b. (1 pt) What is an HSP?
5. Refer to the linked CLUSTALW multiple alignment of a set of GAPDH genes this question. link, CLUSTAL input sequences
- a. (2 pts) Examine the guide tree for this CLUSTAL alignment and list the order the first three the sequences (or sub-alignments) will be aligned to build the final multiple sequence alignment. Your answer should be three pairs of sequences or sub-alignments (i.e., 1. dog/cat 2. [dog/cat]/guppy 3. octopus/cuttlefish).
- b. (2 pt) This CLUSTAL alignment can be improved. Describe two ways you could improve this CLUSTAL alignment and generate a better alignment.
6. The genomic sequence of a hydrogen sulfide producing bacterium from the human oral cavity, Veillonella dispar is sequenced independently by two companies, both to 10X coverage using shotgun paired-end whole genome sequencing. The completed genome is estimated to be 10 Mb. One uses Sanger sequencing and acquires reads with an average length of 900 bp and the second uses an Illumina sequencer and has sequence reads with an average length of 85 bp. As the total sequence is the same there are many more Illumina sequence reads.
- a. (2 pt) Sequence assembly is done independently by both companies. How will the results of the sequence assembly from the Sanger data and the Illumina data differ?
- b. (2 pt) How many gaps do you expect from the Sanger sequencing vs. the Illumina sequencing?
7. (2 pts) In determining the structure of protein using computational methods indicate the type of method appropriate with the circumstance.
Methods: A. Homology modeling, B. Threading, C. Ab initio structure prediction, D. No method likely to work.
- 1. Protein with 80% identity to a protein with an experimentally determined structure.
- 2. Protein with no BLAST match to any protein with an experimentally determined structure.
- 3. Predicted membrane protein, 668 aa, threading fails to find any structure candidates.
8. You wish to construct a phylogenetic tree based on sequences of SRY, the sex determining protein, from a group of equines, donkeys, and zebras.
- a. (1 pt) A SRY protein from what species would be a good choice for rooting the phylogenetic tree?
- b. (1 pt) If after bootstrap analysis a node on the tree has a score of 87%, would you consider the subtree under that node to reliable?
9. (2 pts) List two ways in which the Ensembl gene prediction and annotation system differ from ab initio gene prediction?
10. (2 pts) Microarray transcription analysis allows a biologist to quickly determine the expression level (or relative expression level) of every known gene. There are limitations to microarray analysis. Describe four aspects of gene and protein expression that typical microarrays fail to capture. Number your answers 1 - 4.
11. (3 pts) Describe three things that can be learned from or types of analysis suitable for comparing genomes that have diverged 4 million years (horse-zebra, for example) that can’t be learned from comparing more distantly diverged genomes (mouse-human) or very old splits (tobacco-algae).
12. (2 pt) In a microarray experiment, replicate samples from control and experimental samples are hybridized to arrays. Each group of replicates is going to show variation in gene expression. Normalization tries to minimize the variation beteween samples in a group of replicates so that they can be combined and compared to the experimental groups. Give two sources of variation in gene expression values between sample groups and whether this source of variation will affect some genes, most/all genes, and whether this error is systematic (for example, each gene's expression higher) or random.
13. Refer to the M vs. A graph of yeast spotted microarray data for this question: M_vs_A plot.
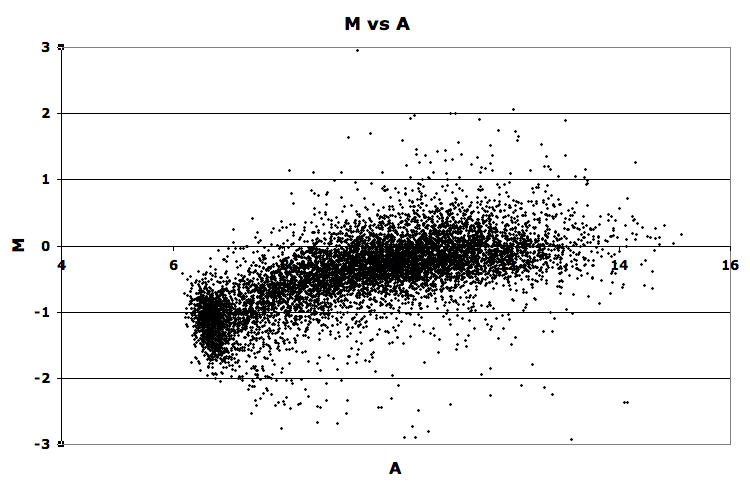{kind=link}
- a. (2 pts). Has this microarray been normalized? Explain the basis for your conclusion.
- b. (2 pts). Give a brief explanation of what 'M' and 'A' represent in the figure.
14. (3 pts) Breast cancer is studied by collecting samples of cancerous tissue from fifty women and normal breast tissue from fifty control women of similar age, race, and health. These samples are hybridized to Affymetrix full genome microarrays. One approach for analyzing this experiment is to pick the 2,000 genes that vary the most in the samples and perform a heirarchical clustering, clustering both genes and samples using Pearson correlation (center) as the similarity measure. What could you learn about breast cancer from this analysis? Describe three clusters of genes and/or breast tissue samples that might be observed and what they would indicate.
15. (2 pts) One problem with using protein interaction databases such as IntAct is that high throughput studies utilizing yeast 2-hybrid or proteomics techniques tend to have a high false positive rate. You are looking for proteins that interact with Map kinase. Describe bioinformatic methods of weeding out false positives from the initial search results, or separating proteins you can be confident interact with Map kinase of from lower confidence results.
16. (2 pts) Examine the region near SOX21 locus, expand the browser region to a 10-20kb region around the gene. What is unusual about the region 5' and 3' (especially 3') of SOX21?
17. (2 pts). Aside from its sequence what other information describing a SNP is the most important and useful to know?
18. (12 pts) Fill in the blank with one, two, or (rarely) three words.
a. The NCBI __________ sequence database is a primary, repository database.
b. The ________________ is a measure of the likelihood of finding an HSP with the given score in a sequence database calculated by the BLAST program.
c. __________________ are used to compensate for zero counts (i.e., missing values) when calculating scores in a PSSM.
d. Each cistron in a multi-gene operon has its own ___________________ signal sequence.
e. Eukaryotic genes with _________________ bp composision are often missed by de novo gene prediction programs.
f. An advantage of NMR is that __________________ are determined and they can provide insight into protein dynamics.
g. Finding overlapping residues in an experimentally determined protein structure is __________________.
h. In an RNA structure, the most confidently predicted parts of a secondary structure are bp regions with __________________ predicted pairing.
i. The maximum Likelihood (ML) algorithm is a __________________ based method and thus is slower than the Neighbor Joining method.
j. _____________________ is used to assess support for nodes in a phylogenetic tree.
k. In ___________________ genome sequencing, millions of short DNA sequences are determined simultaneously.
l. In genome sequencing, _________________ is the total number of bps sequenced divided by the total genome size.