The purpose of this lab is to familiarize you with two types of alignments:
-
one-to-one
-
one-to-database
This familiarity should encompass several aspects:
-
The basis of alignment methods.
-
Commonly used, flexible tools for alignment.
-
Interpretation of alignments.
-
Relating alignments to biological structure and function.
E-mail your lab to the class TA, Yeshi (tgyeshi@uky.edu) with a subject line "BIO520 Lab 3". Use Rich Text Format (.rtf) or MS Word Format (.doc) and name the document like so: LundJ_lab3.rtf. This lab is due Thurs 2/4/2010.
There is NO bioinformatic method more important or more used than alignment...learning this well will make you a better biologist.
Alignment Methods
Alignment is an assertion of the similarity of two sequences. The result of such a sequence comparison depends upon the criteria for scoring a match and upon the algorithm for finding a match. Note that biologists often assume that there is a single "correct" alignment, when in fact there are many possible alignments and several alternatives may have particular statistical and biological significance. The statistical significance of one alignment compared to another alignment is one important way to assess the value of the alignment, but even statistics cannot be a perfect arbiter of value. And, finally one must realize that biological significance of a particular alignment is usually the reason for which the alignment was performed.
NCBI BLAST
1. Use the DNA scoring matrix below (the default BLAST nucleotide matrix):
| Score | |
| Identity | +2 |
| Mismatch | -3 |
| Gap creation | -5 |
| Gap extension | -2 |
| Terminal Mismatch | 0 |
a. Score the following DNA sequence alignment:
TCCTAGGACTCATCGTAAGG
TCCTAGAACCTCGTAAGG
ANSWER=score
b. I think it is easy to find an optimal alignment for these two sequences with this matrix. Please paste in the ALIGNMENT in PROPER FORMAT (non-proportional font).
c. Compute the new, optimal score ANSWER=score.
2. I have used the following DNA sequence in Dotlet to generate a dotplot of the sequence to itself. Please explain the following observations:
|
a. Why is there a perfect diagonal (labeled a)? b. What does the element labeled b derive from (there are a few of these, actually)? c. What is a biological meaning or interpretation of these sequences? (Note how difficult it would be to find these regions using normal pairwise tools.) |
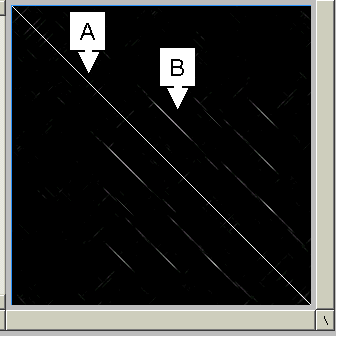 |
3. Please align the two nexin polypeptide sequences from human (Q9Y5X1) and zebrafish (AAI67965.1). Use the BLAST 2 program (go to 'Align two (or more) sequences using BLAST (bl2seq)') to align the polypeptides over their length (all defaults, make sure the 'blastp' program is selected).
a. Report % identity, length of match.
b. By default, BLASTP uses the BLOSUM62 matrix. How does the alignment score change when the BLOSUM80 matrix is used?
Now use BLAST 2 to align the coresponding human (AF121859) and zebrafish (BC167965.1) nexin DNA sequences. Make sure 'blastn' is selected under 'Program Selection'.
c. Report % identity, length of *each* match (HSP).
d. Explain the differnces seen between the protein and DNA aligments in 3c and 3a. Write a ONE SENTENCE explanation of why there is this difference between the protein and DNA alignment?
e. The 3' ends of the two DNA sequences do not match. Write ONE SENTENCE in which you explain why this is?
4. BLAST programs and finding matches.
You have sequenced a random segment of genomic DNA from Echinops telfairi (given in FASTA format). You would like to know whether a segment of DNA overlapping yours (identical match) has been sequenced.
a. Which combinations of NCBI BLAST programs and databases would you use for this determination.
Please give your answer like this:
LIST the program and databases like so: blastp/nr AND blastp/swissprot...etc.
b. You would like to know whether it is possible that this segment of DNA
is part of an exon that encodes a protein similar to any already-sequenced, publicly accessible
sequence. List the blast programs and databases that you feel would EXHAUSTIVELY address the question of relatedness.
5. You want to find genomic sequence around human insulin-like growth factor 1 receptor (IGF1R, Accession number X04434). Use megablast to do this.
a. What database do you search?
b. Where is this gene located?
c. How many exons does the X04434 sequence contain? There are a couple of ways to determine this. One way is to click on 'Formatting options', then slect the checkbox 'Use old BLAST report format', and select the 'Alignment View' of 'Hit Table' may help. Also, one of the searches gives you a 'Genome Alignment' view option which is useful.
6. In a yeast 2-hybrid screen of a Xenopus laevis cDNA library you identify a clone that binds to ZNF74 and have it sequenced. Use BLAST to try to identify the sequence.
a. This sequence has two regions of similarity. Briefly describe what each region matches.
b. What is this cDNA? ANSWER=a few words.
7. Use the sequence of a metabolic enzyme (5,10-methylene tetrahydrofolate dehydrogenase-the folD gene product) from E. coli to find polypeptides that are close relatives. This protein has been sequenced from many organisms and you will need to increase the 'Max target sequences' from the default values. Increase these values until the list of matches includes some poor matches. In viewing the results you may need to Reformat to show more alignments.
a. Give the NAME of the database search
program and the NAME of the database most appropriate for this task.
Run this search.
b. This BLAST program also searches the conserved domain database (CDD) and indicates domains below the match. Give the domains found and their location in the E. coli protein. Answer like this: FOO 10-44aa, GLOBIN 82-122aa.
b. List the gi numbers for the most similar polypeptide found in Arabidopsis thaliana and at least one gi number for the most similar polypeptide found in Saccharomyces cerevisiae. There are several ways of finding this information; you can scan the standard BLAST report, use the 'Taxonomy reports' format, or limit your BLAST search by organism.
c. How many human homologs of this E. coli gene are there? What is the Score and E-value of the most distant homolog (the worst match that you think is meaningful and due to homology rather than chance).