BIO520 Exam 2 Spring 2009
Please email this lab to Jim Lund (jiml@uky.edu) with a subject line "BIO520 Exam 2" and name the document like so: "LundJ_exam1" or hand in written answers. Fill in your name on the exam!
You may use any books, notes, web pages, software programs, or related materials to complete this exam. You MAY NOT consult with any person regarding the exams intellectual content.
1. Gene find in prokaryotes and eukaryotes.
- a. (3 pt) Name three criteria, sequence features, sequence properties, or algorthms used for gene finding in both prokaryotic and eukaryotic genomic DNA.
- b. (2 pt) Why is gene finding easier in prokaryotes than in vertebrate genomes?
- c. (3 pt) Name three criteria, sequence features, sequence properties, or algorthms used specifically in eukaryotic gene finding.
2. Answer the following questions based on the single Genscan predicted exon shown below:
Gn.Ex Type S .Begin . .End .Len Fr Ph I/Ac Do/T CodRg P. . . Tscr. .
----- ---- - ------ ------ ---- -- -- ---- ---- ----- ------ -------
7.03 Term + 34524 34652 129 2 0 101 50 83 0.373 3.00
- a. (1 pt) How many bps long is this exon?
- b. (1 pt) Is this exon predicted to be on the parallel or the anti-parallel strand?
- c. (1 pt) How confident is Genscan in its prediction of this exon?
- d. (1 pt) What type of an exon is this?
3. Assessing gene finding.
- a. (1 pt) How well does de novo gene finding work?
- c. (1 pt) Why is gene finding such a difficult problem?
4. Rubisco is the plant enzyme that takes CO2 and attaches it to ribulose bisphosphate, and then clips the result into two phosphoglycerate molecules. Examine the PDB entry for the rubisco protein from spinach. PDB link, 1rcx.pdb.
- a. (1 pt) How many protein chains are present in this structure?
- b. (1 pt) What is the resolution of this structure? Include the appropriate units in your answer.
- c. (1 pt) Which molecules other than protein chains are present in the structure?
- d. (2 pt) Examine the Ramachandran plot for this structure: 1RLC_Rama.pdf. What does the plot tell you about the quality of the structure? Explain your answer.
5. (3 pts) Given a protein with homology only to other proteins of undertermined function, describe steps you could take to characterize it computationally. Give two things you attempt to predict about it and the program/analysis you would use.
6. Secondary structure of human SIRT1. Examine the STR4 secondary structure predictions for this protein: sequence logo.
- a. (2 pt) What secondary structure is predicted at aa 30-38 (line 1, 'CHKFIALSD')? At aa 140-144 (line 2 'LLIVI')?
- b. (1 pt) Overall, would you describe the STR4 SIRT1 secondary structure predictions as having high or low confidence? Briefly describe your reasoning.
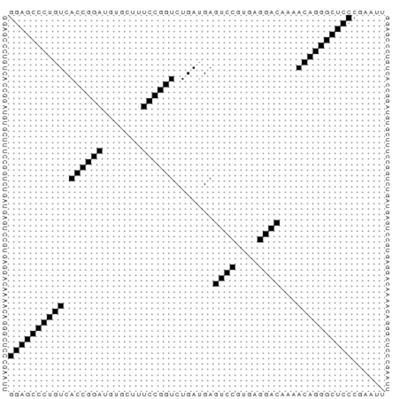
7. (2 pts) Examine the dotplot of pairing for RNA sequence 'ggagccctgtcaccggatgtgctttccggtctgatgagtccgtgaggacaaaacagggctcccgaatt' shown here RNA dotplot. Base pair 1 is at the top left, bp 68 is at the bottom right. On the provided piece of paper 1) write your name and 2) draw this RNA structure.
8. (8 pts) Match each left column database/program with a single application in the right column. There are extra entries in the right column. Inidcate your answer as (A8, B10, C4, etc).
A. GeneMark
B. PHYLIP
C. PDB
D. TFD
E. JPRED
F. TargetP
G. PSORT II
H. JMol
I. Cn3D
J. MFOLD
K. WoLF PSORT
L. ESyPred3D
M. TopPred
N. JalView
O. GENSCAN
P. PDB
0. Predict membrane topology of proteins with membrane spanning α-helices.
1. Predict membrane topology of proteins with membrane spanning β-strands.
2. Protein threading server.
3. Manipulate sequences and formats, annotate seqeunces.
4. Uses homology moldeling to predict protein structure.
5. Protein subcellular location.
6. DNA sequencing and assembly.
7.Protein structure database.
8. Phylogenetics trees construction and analysis.
9. Transcription factor binding site database.
10. NCBI's protein structure viewer.
11. Web browser protein structure viewer
12. RNA folding.
13. Protein structure database.
14. RNA structure database.
15. De novo gene finding
16. Predict exon-intron gene structure in genomic DNA.
17. Predict pretein secondary structure.
18. Subcellular localization prediction for eukaryotic proteins.
19. Subcellular localization prediction using signal sequences and homology.
20. Edit multiple sequence alignments.
8. Phylogenetics
- a. (2 pt) What is the difference between a phylogram and a cladogram?
- b. (2 pt) Explain one method of rooting a phylogenetic tree.
- c. (2 pt) When comparing orthologous protein sequences from two species is the number of aa changes directly proportional to the time the two species diverged? Briefly explain why or why not.
- d. (2 pt) The maximum parsimony method for building phylogenetic trees is a character based method that used an optimality criteria to choose the best tree. How does a character based method differ from a distance based method with respect to tree construction and time required?
- e. (1 pt) Bootstrap analysis is run on a phylogenetic tree using 1000 trials. Give a bootstrap value that would indicate a strongly supported clade.
9. (1 pt) Which is more strongly conserved, protein primary, secondary, or tertiary structure?
10. (3 pts) You construct a phylogenetic tree of fire ants and twelve other ant species from mitochondrial DNA sequences using the minimal evolution method while another graduate student in your lab uses the same sequences and the UPGMA method. Your trees differ. How can you resolve this problem and find a tree in which you are confident?
BIO520
|
{kind=link}