BIO520 Exam 2 Spring 2010
Please email this lab to Jim Lund (jiml@uky.edu) with a subject line "BIO520 Exam 2" and name the document like so: "LundJ_exam2" or hand in written answers. Fill in your name on the exam!
You may use any books, notes, web pages, software programs, or related materials to complete this exam. You MAY NOT consult with any person regarding the exams intellectual content.
1. Examine the PDB entry 1W0E for protein CYP3A4.
- a. (1 pt) What method was used to determine this structure, and what is its resolution?
- b. (1 pt) This structure has an R-value of 0.244. What is the R-value?
- c. (1 pt) Is this resolution sufficient to resolve only the protein backbone, individual residues, or atoms?
- d. (1 pt) What is the large molecule in the center of the protein?
- e. (1 pt) Is this protein primarily alpha helix, beta sheet, or mixed?
- f. (1 pt) Along its longest dimension, what is the diameter of one subunit of this protein? Include units in your answer.
3. (2 pts) In determining the structure of protein using computational methods indicate the type of method appropriate with the circumstance.
Methods: A. Homology modeling, B. Threading, C. Ab initio structure prediction, D. No method likely to work.
- 1. Protein with 80% identity to a protein with an experimentally determined structure.
- 2. Protein with no BLAST match to any protein with an experimentally determined structure.
- 3. Predicted membrane protein, 668 aa, threading fails to find any structure candidates.
4. (2 pts) Given a protein with homology only to other proteins of undertermined function, describe steps you could take to characterize it computationally. Give two things you attempt to predict about it and the program/analysis you would use.
5. (2 pts) Examine the energy plot of pairing for RNA sequence 'gcgtgcggctatgtgctagagggcaaagagcttcggttctatcttaggaaatgaagggcccgtggcacaagcgccgcaa' shown here: RNA dotplot. Base pair 1 is at the top left, bp 79 is at the bottom right. On the provided piece of paper 1) write your name and 2) draw this RNA structure. Ignore the sub-optimal lower energy matches (the colored sections).
6. The genomic sequence of a hydrogen sulfide producing bacterium from the human oral cavity, Veillonella dispar is sequenced independently by two companies, both to 10X coverage using shotgun paired-end whole genome sequencing. The completed genome is estimated to be 10 Mb. One uses Sanger sequencing and acquires reads with an average length of 900 bp and the second uses an Illumina sequencer and has sequence reads with an average length of 85 bp. As the total sequence is the same there are many more Illumina sequence reads.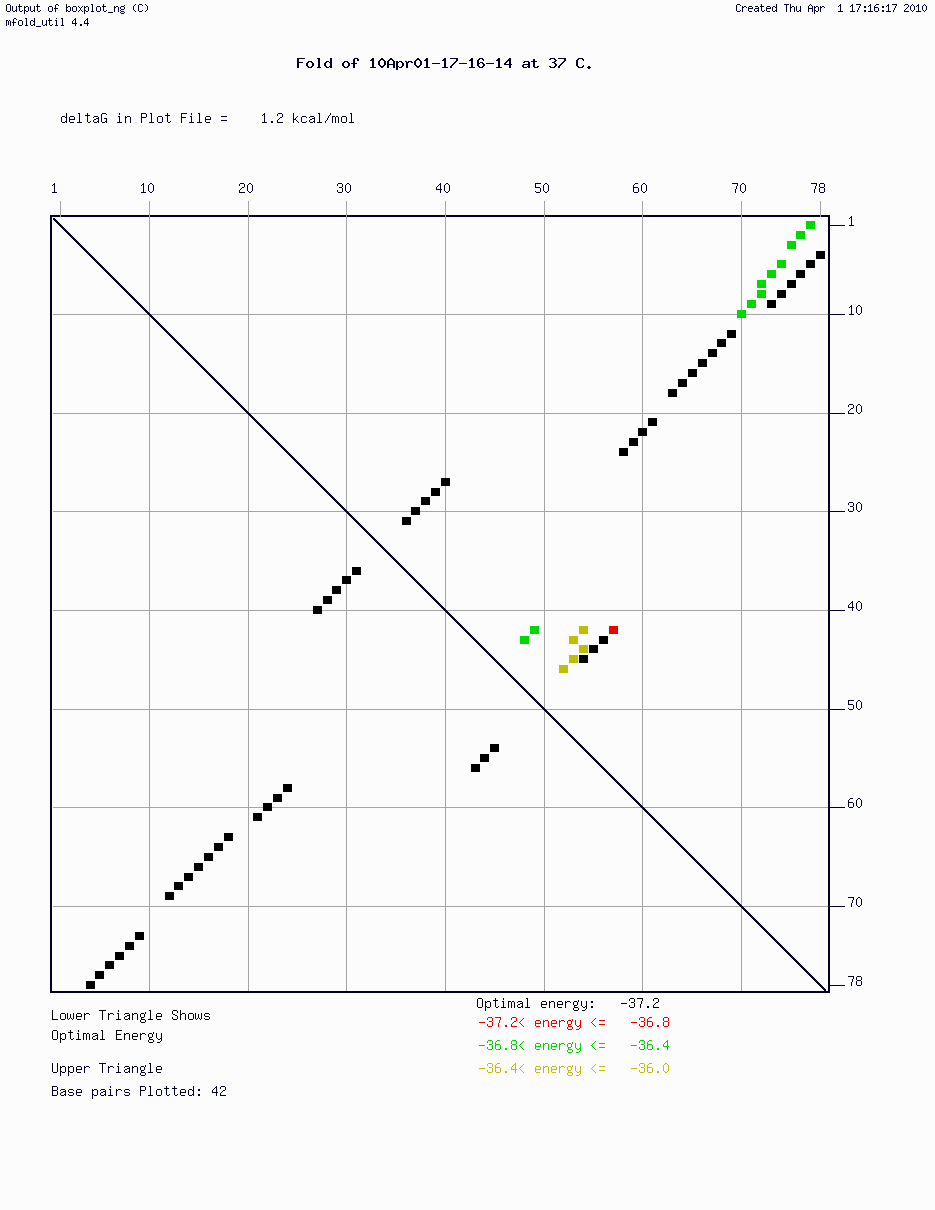{kind=link}
- b. (2 pt) Sequence assembly is done independently by both companies. How will the results of the sequence assembly from the Sanger data and the Illumina data differ?
- a. (2 pt) How many gaps do you expect from the Sanger sequencing vs. the Illumina sequencing?
7. (2 pt) The DOE sequencing finishing standards specify 99.9% per base accuracy. Briefly describe two serious problems that would arise from working with genomic sequence with a 99% accuracy, an average of one error in 100 bp.
8. Examine the Genscan gene predictions for a C. familiaris genomic sequence, Genscan output.- a. (3 pt) For each predicted gene, give the number of exons and the strand.
- b. (1 pt) Does Genscan rate these predictions as high or low confidence?
- c. (2 pt) These predictions correspond poorly to the NCBI genome annotation made with GNOMON. What other criteria/methods (other than de novo gene prediction) are/can be used be GNOMON to improve gene and exon prediction?
- a. (3 pt) Name three criteria, sequence features, sequence properties, or algorithms used for gene finding in both prokaryotic and eukaryotic genomic DNA.
- b. (2 pt) Why is gene finding easier in prokaryotes than in vertebrate genomes?
A. GeneMark B. VAST C. PDB D. TRANSFAC E. JPRED F. TargetP G. PSORT II H. JMol I. Cn3D J. MFOLD K. WoLF PSORT L. Artemis M. Phrap N. Phyre O. PDB
0. Predict membrane topology of proteins with membrane spanning α-helices. 1. Predict membrane topology of proteins with membrane spanning β-strands. 2. Protein threading server. 3. Program to assemble contigs from DNA sequences. 4. Uses homology modeling to predict protein structure. 5. Protein subcellular location. 6. DNA sequencing and assembly machine. 7. Protein structure database. 8. Phylogenetics trees--construction and analysis. 9. Transcription factor binding site database. 10. NCBI's protein structure viewer. 11. Web browser protein structure viewer 12. RNA folding. 13. Protein structure prediction web server. 14. RNA structure database. 15. De novo gene finding 16. Predict exon-intron gene structure in genomic DNA. 17. Predict protein secondary structure. 18. Subcellular localization prediction for eukaryotic proteins. 19. Subcellular localization prediction using signal sequences and homology. 20. Edit multiple sequence alignments. 21. Program for aligning protein structures. 22. View, edit, and analyze DNA annotations of genes and other sequence features.